
Randy Wadkins, Professor of Chemistry & Biochemistry
Professor of Chemistry & Biochemistry
409 Coulter Hall
662-915-7732 | rwadkins@ olemiss.edu
EDUCATIONAL AND PROFESSIONAL BACKGROUND
B.S., University of Mississippi, 1986
Ph.D., University of Mississippi, 1990
NIH Predoctoral Fellowship, University of Mississippi, 1986-1990
Max Plank Gesellschaft Postdoctoral Fellow, Max Planck Institute for Biophysical Chemistry, Goettingen, Germany, 1990-1991
Postdoctoral Fellowship, St. Jude Children’s Research Hospital, 1991-1994
AAAS S&T Congressional Fellow 2015-2016
PROFESSIONAL RECOGNITION
Biophysical Society Congressional Fellowship
RESEARCH INTERESTS
Biophysical chemistry, molecular dynamics, fluorescence microscopy and imaging, DNA structure and structural transitions, biosensors
RESEARCH SUMMARY
The primary focus of my lab is focused on developing improved antitumor drugs of the camptothecin family. Camptothecins are among the most promising new antitumor agents available. We are working on a “third generation” of camptothecins that are superior in activity to the clinically-used 2nd generation agents CPT-11 (Irinotecan) and topotecan (Hycamtin) just as these agents are superior to the parent camptothecin.
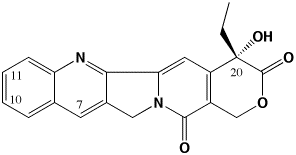
Fig. 1. The molecular structure of camptothecin
The 3rd generation of camptothecins are being tested for their ability to induce topoisomerase I-DNA complexes, and importantly, to induce temporally stable complexes. Camptothecins are specific topoisomerase I-targeting antitumor drugs. They are S-phase selective due to the need for DNA replication in their killing mechanism. However, camptothecins induce DNA damage in all phases of the cell cycle. We are developing analogs that can induce long-lasting DNA damage in G0/G1 phase of the cell cycle such that lethality ensues once these cells enter S-phase. Such analogs may be more effective in slow-growing tumors such as prostate and pancreas where there is presently no effective chemotherapeutic treatment. To develop these compounds, we are measuring the kinetics of dissociation of topoisomerase I from DNA as a function of chemical substitution on the camptothecin pharmacophore. This methodology reveals which substituents are able to induce long-lived complexes. We have recently demonstrated that the present 2nd generation of camptothecins may be limited in their activity because they form short-lived complexes. We use a variety of biophysical techniques to probe the interaction of camptothecins with topoisomerase I and DNA, including fluorescence imaging microscopy and temperature and osmotic pressure effects on rate constants for the dissociation of camptothecins from the topo I-DNA complex. We are now developing spectroscopic techniques to monitor drug, topoisomerase I, and DNA interactions in real (microsecond) time to characterize more clearly the way in which these drugs interact with their target. We have also recently determined that water participation is an important component of these interactions.
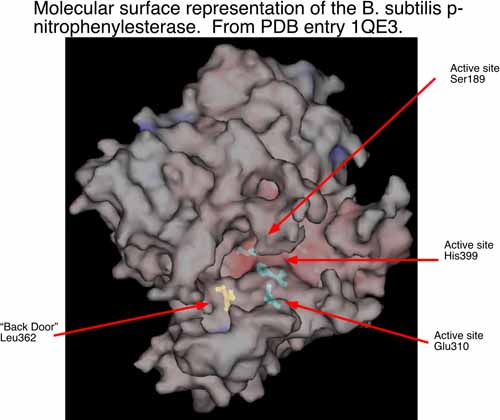
Figure 2: The active site of the B. subtilis enzyme that activates CPT-11. p-nitrophenylesterase
The second area of research in my laboratory is in the interface of experimental and computational chemistry. We have began using computational structural biology to interpret and predict experimental results in biochemistry. In collaboration with Dr. Philip Potter at St. Jude Children’s Research Hospital (Memphis, TN), we are currently using computational methods to understand the conversion of CPT-11 to SN-38 by liver carboxylesterases and how this process might be enhanced by selecting analogs of CPT-11 specific to selected carboxylesterases. The collaboration with Dr. Potter has identified a bacterial counterpart to the mammalian enzymes examined previously. The p-nitrophenylesterase from B. subtilis efficiently hydrolyzes CPT-11 and it can be readily mutated for analysis of specific residues that contribute to conversion of CPT-11. This enzyme has also been solved by x-ray crystallography. We are currently running molecular dynamics simulations of this enzyme and two mutants known to affect CPT-11 conversion by the enzyme. Hence, we have a unique opportunity to examine the molecular underpinnings of substrate selectivity across evolutionary divergence.
A third research interest is in DNA secondary structures as likely targets for drug development. Targeting drugs to select DNA sequences has been of interest to many investigators for years. However, in practice it is difficult to target long sequences of DNA due to the repetitive conformation of the DNA major and minor grooves. One way to target longer sequences of DNA is if these sequences adopt higher order (or “secondary”) structures, including DNA hairpins and their cousins the quadraplexes (or G-tetraplexes). Although these secondary structures were originally implicated in telomere maintenance, they have recently been demonstrated to have regulatory roles in the expression of a number of genes. These include transcription of β-casein, human platelet-derived growth factor, epidermal growth factor, c-myc, and human enkephalin. I have reviewed this topic in Wadkins (2000) Curr. Med. Chem. 7: 1-15. Figure 3: How small molecules may interfere with DNA secondary structures. In (A), duplex DNA may open spontaneously or may be opened by DNA-binding proteins to allow instigation of transcription. In (B), small DNA-binding molecules interfere with this process. DNA secondary structure Work from my colleagues Drs. Von Hoff and Laurence Hurley at the Arizona Cancer Center have recently demonstrated that c-myc can be effectively silenced by quadraplex-interactive drugs. This opens a great number of doors for the development of drugs that interact with these structures. At present, my lab is purifying a yeast transcription factor, RAP1, that is known to promote quadraplex formation in single-stranded DNA. Our goal is to use this factor to probe for quadraplex structure in a number of promoters for genes involved in cancer drug resistance, and to develop compounds that interfere with its binding. Further, following from the work of Craig Benham at UC-Davis, we are interested in how RAP1 may influence the ability of supercoiling in DNA to cause extrusion of these secondary structures.
The last area I am working in is the use of biosensing surfaces for the detection of cancer. Our work in this area has focused on the development of an implantable, optical fiber-based probe for detection of circulating tumor cells. Our sensor is based on a modified optical waveguide probe that detects signals from a fluorescent dye bound in proximity to its surface. Tumor cell attachment to the surface of the probe initiates a signal from the fluorescent dye that is detected by the device. This signal is then propagated to external electronics to indicate the presence of circulating tumor cells. The general description of such sensors is given in our review: Wadkins and Ligler (1998) Meth. Biotechnol. 7: 77. To construct the biosensor, we are developing a functionalized Polyethylene oxide (PEO)/Polystyrene (PS) surface that specifically binds cancer cells in vitro. We are producing an optical probe from the ligand-modified PEO/PS polymer that is suitable for implantation in the body. Finally, we are using this optical sensor for detecting and reporting binding of circulating cancer cells. Figure 4: Fiber optic biosensor
RECENT PUBLICATIONS
Complete citations for Randy M. Wadkins (link to NCBI database) Search for RM Wadkins